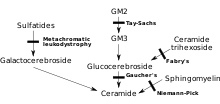
Sphingolipidoses are a class of lipid storage disorders or degenerative storage disorders caused by deficiency of an enzyme that is required for the catabolism of lipids that contain ceramide,[1] also relating to sphingolipid metabolism. The main members of this group are Niemann–Pick disease, Fabry disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and metachromatic leukodystrophy. They are generally inherited in an autosomal recessive fashion, but notably Fabry disease is X-linked recessive. Taken together, sphingolipidoses have an incidence of approximately 1 in 10,000, but substantially more in certain populations such as Ashkenazi Jews. Enzyme replacement therapy is available to treat mainly Fabry disease and Gaucher disease, and people with these types of sphingolipidoses may live well into adulthood. The other types are generally fatal by age 1 to 5 years for infantile forms, but progression may be mild for juvenile- or adult-onset forms.
^Lynn, D. Joanne, Newton, Herbert B. and Rae-Grant, Alexander D. eds. 5-Minute Neurology Consult, The. 2nd Edition. Two Commerce Square, 2001 Market Street, Philadelphia, PA 19103 USA: Lippincott Williams & Wilkins, 2012. Books@Ovid. Web. 03 December, 2020
Sphingolipidoses are a class of lipid storage disorders or degenerative storage disorders caused by deficiency of an enzyme that is required for the catabolism...
play important roles in signal transduction and cell recognition. Sphingolipidoses, or disorders of sphingolipid metabolism, have particular impact on...
(including Tay–Sachs disease (E75.0-E75.1) - they are a subtype of sphingolipidosesSphingolipidoses that are not gangliosidoses, including Gaucher's and Niemann–Pick...
glycans from glycolipids to turn them back into unmodified lipids. Sphingolipidoses are a group of diseases that are associated with the accumulation of...
Many lipid storage disorders can be classified into the subgroup of sphingolipidoses, as they relate to sphingolipid metabolism. Members of this group include...
then solely formed via degradation of sphingolipid in the lysosome. Sphingolipidoses General structures of sphingolipids Dimethylsphingosine Fingolimod...
from necrosis and apoptosis occurring after acute spinal cord injury. Sphingolipidoses Structures of GM1, GM2, GM3 gangliosides Mocchetti I (2005). "Exogenous...
from the similarity in presentation to both mucopolysaccharidoses and sphingolipidoses. A biochemical understanding of these conditions has changed how they...
commonly listed in the family of leukodystrophies as well as among the sphingolipidoses as it affects the metabolism of sphingolipids. Leukodystrophies affect...
accumulation of GM1 gangliosides. They are inherited, autosomal recessive sphingolipidoses, a class of lipid storage disorders. Diagnosis of GM1 can be obtained...
pathology and ultrastructure and also different from other forms of sphingolipidoses.[citation needed] Subsequently, Santavuori and Haltia showed that an...
and multiple sulfatase deficiency are classified as sulfatidoses. Sphingolipidoses#Overview for an overview table, including sulfatidosis "Definition:...
glycogenosis type II, storage diseases (Gaucher, Niemann Pick and other sphingolipidoses) mitochondrial diseases and genetic immunodeficiencies such as IPEX...
 Global Information
Global Information